Abstract by Marie Elodie Hélène Cadot
Targeting the protein-protein interaction between Keap1 and Nrf2 is a promising strategy against oxidative stress and inflammation-related pathologies. Several covalent inhibitors that activate Nrf2 have been approved in the past decades. However, their irreversible and unspecific binding can cause off-target effects. To address this, noncovalent small-molecule Keap1-Nrf2 inhibitors are being developed, offering a promising alternative due to their improved selectivity. To date, none has received regulatory approval.
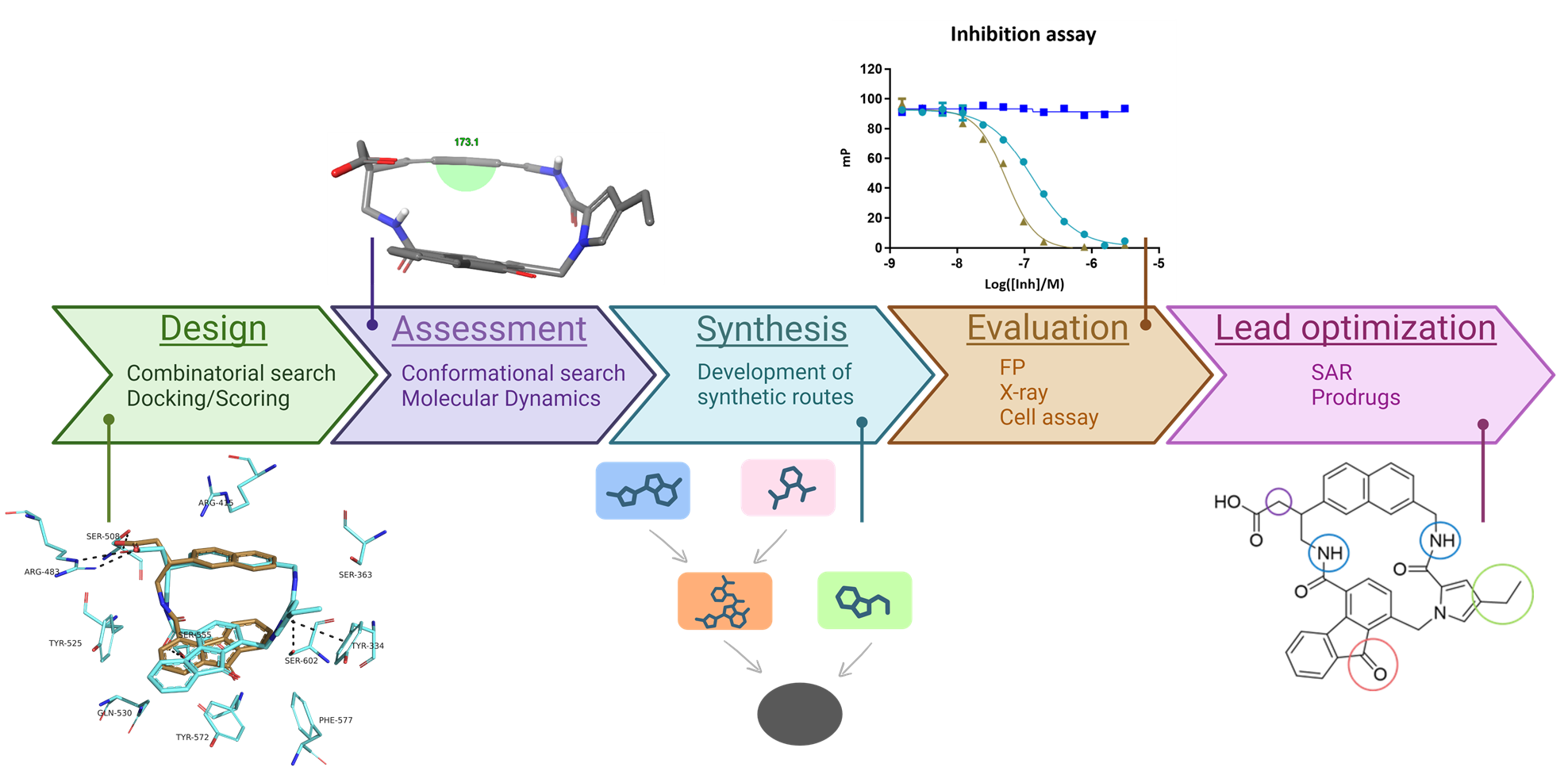
We have previously designed noncovalent Keap1-Nrf2 inhibitors showing moderate to high affinities towards Keap1. However, the structures include carboxylic acids, which can reduce membrane permeability. Here, we explored a macrocyclic series as an attempt to overcome this issue, as macrocycles have been described to enhance affinity, selectivity, stability and sometimes permeability.
We designed and evaluated 108 macrocycles in silico, using molecular docking, conformational analysis and molecular dynamics. Three macrocyclic structures were prioritized for synthesis. After intensive synthetic efforts, two macrocycles (MC47 and MC100) were obtained and evaluated for Keap1-Nrf2 inhibition in fluorescence polarization assay, showing affinities of 14 and 40 nM. An X-ray structure of MC100 in complex with the Keap1 Kelch domain was elucidated, which validated our docking results and computational assessment. Hence, lead optimization was initiated to improve the physicochemical properties of the molecule, applying a structure-activity relationship study to develop analogues of MC47. Moderate activity was observed in cells for some macrocycles, correlating with key physicochemical properties (cLogD, tPSA). A prodrug strategy was also investigated for one compound but did not offer improvement in cells.